Gonadal Dysgenesis in 46,XY Females (XY Gender-Reversal)
Authors
INTRODUCTION
Gonadal dysgenesis may occur in individuals with apparently normal male (46,XY) chromosomal complements, and the phenotype may be indistinguishable from 46,XX gonadal dysgenesis with normal stature. Actually, this is entirely predictable because loss of testicular tissue before 7–8 weeks of embryogenesis was shown half a century ago by Jost1 to produce such a phenotype in rabbits. The spectrum of the disorders of gonadal dysgenesis is shown in Table 1, and in this chapter these disorders are systematically reviewed. Related chapters include "Genetics of Sexual Differentiation", in which Ostrer reviews the genetics of testicular differentiation and "XX Gonadal Dysgenesis and Premature Ovarian Failure (POF) in 46,XX Individuals", in which Simpson reviews gonadal failure in 46,XX women is reviewed. This chapter inevitably reflects the author’s other publications on the topic.2, 3
TABLE 1. The spectrum of 46,XY gender reversal (XY females)
XY gonadal dysgenesis without somatic anomalies
Perturbations of SRY (HMG-box)
Duplication Xp (DAX1)
X-linked recessive form
Forms without detectable molecular perturbation or heritability
XY gonadal dysgenesis and Wilms’ tumor oncogene (WT1)
Denys-Drash syndrome
Frasier syndrome
XY gonadal dysgenesis with malformation
XY gonadal dysgenesis and campomelic dysplasia (SOX9)
XY gonadal dysgenesis and α-thalassemia X chromosome (ATX) syndrome
XY gonadal dysgenesis in other malformation syndromes
Ectodermal anomalies (Brosnan)
Genital-palato-cardiac (Gardner-Silengo-Wachtel)
Spastic paraplegia-optic atrophy-microcephaly (Teebi)
XY gonadal dysgenesis with autosomal deletions
Del (2p)
Del (9p)
Del (10q)
XY gonadal dysgenesis with autosomal duplications
Dup1p (WNT1)
Dup17q (SOX9)
Germ-cell failure in both sexes (46,XY cases)
No somatic anomalies
Hypertension and deafness
Alopecia
Microcephaly and short stature
Leydig cell hypoplasia
Steroid biosynthetic defects
Steroidogenic factor 1 deficiency (StAR)
17α-hydroxylase/17,20-desmolase deficiency (CYP17)
3β-ol dehydrogenase/3β-hydroxysteroid dehydrogenase deficiency (3βHSD)
Agonadia (46,XY)
REPRODUCTIVE EMBRYOLOGY
Primordial germ cells originate in the endoderm of the yolk sac and migrate to the genital ridge to form the indifferent gonad. 46,XY and 46,XX gonads are initially indistinguishable. Indifferent gonads develop into testes if the embryo, or more specifically the gonadal stroma, is 46,XY (Fig. 1). This process begins approximately 43 days after conception. Testes become morphologically identifiable 7–8 weeks after conception (9–10 gestational or menstrual weeks).3
Sertoli cells are the first cells to become recognizable in testicular differentiation. These cells organize the surrounding cells into tubules. Both Leydig cells4 and Sertoli cells5 function in dissociation from testicular morphogenesis, consistent with these cells directing gonadal development rather than the converse. These two cells secrete hormones that direct subsequent male differentiation (see Fig. 1).
|

Fetal Leydig cells produce an androgen, testosterone, that stabilizes wolffian ducts and permits differentiation of vasa deferentia, epididymides, and seminal vesicles. After conversion by 5α-reductase to dihydrotestosterone (DHT), external genitalia are virilized. These actions can be mimicked by the administration of testosterone to female or castrated male embryos, as demonstrated clinically by the existence of teratogenic forms of female pseudohermaphroditism.
Fetal Sertoli cells produce antimüllerian hormone (AMH), also known as müllerian inhibitory substance (MIS). This glycoprotein diffuses locally to cause regression of müllerian derivatives (uterus and fallopian tubes). When AMH is chronically expressed in XX transgenic mice, oocytes fail to persist, tubule-like structures develop in gonads, and müllerian differentiation is abnormal.6 Thus, AMH may have functions related to gonadal development as well. Steroidogenic factor 1 (SF-1) appears to regulate AMH, given that a uterus was present in a gender-reversed XY female lacking SF-1.7
In the absence of a Y chromosome, the indifferent gonad develops into an ovary. Transformation into fetal ovaries begins at 50–55 days of embryonic development. Whether female (ovarian) differentiation is truly a default (constitutive) pathway or whether a yet to be determined gene product directs primary ovarian differentiation is uncertain. That 46,XY individuals with certain syndromes (Gardner-Silengo-Wachtel) may show oocytes, as may XY gonadal dysgenesis neonates,8 favors the default hypothesis. Partial ovarian function was also found in a gender-reversed XY case having a de novo mutation (Gln2Stop) at the 5` end of SRY.9 Relevant is that germ cells are present in 45,X embryos,10 only to undergo atresia at a rate more rapid than that occurring in normal 46,XX embryos; thus, pathogenesis involves not only failure of germ cell formation but also increased atresia. Perhaps the same phenomenon occurs in XY gonadal dysgenesis.
Internal ductal and external genital development is secondary to but independent of gonadal differentiation. In the absence of testosterone and AMH, external genitalia develop in female fashion. Müllerian ducts form the uterus and fallopian tubes, and wolffian ducts regress. This scenario occurs in normal XX embryos as well as in XY embryos (animals) castrated before testicular differentiation.
XY GONADAL DYSGENESIS (SWYER SYNDROME)
The prototypic example of the XY female has gonadal dysgenesis but structurally normal female external genitalia, vagina, uterus, and fallopian tubes. In at least some cases the gonads of human XY females are embryologically ovaries.11 Secondary sexual development does not occur at puberty. Height is normal and somatic anomalies are usually not present. This external appearance may be identical clinically to that of 46,XX gonadal dysgenesis without somatic anomalies. Gonadotropins (follicle-stimulating hormone [FSH], luteinizing hormone [LH]) are elevated and estrogens decreased. Hormonal treatment is similar to that used for any patient with primary gonadal failure. XY gonadal dysgenesis was once called Swyer syndrome, after the initial description of Swyer and colleagues12 of a normal-stature X-chromatin-negative patient of female appearance and having streak gonads.12
An important clinical feature is the high risk of dysgerminoma or gonadoblastoma, which is approximately 20–30% without medical intervention.13 Neoplasia may even arise in the first or second decade. Any XY female experiencing secondary sexual development (breasts, pubic hair) should be suspected of doing so because of hormone-producing neoplasia (dysgerminoma, gonadablastoma). The relatively high likelihood of undergoing neoplastic transformation necessitates gonads being extirpated in XY gonadal dysgenesis. Laparoscopic removal of gonads and sometimes tumors is possible (Fig. 2).14, 15 The uterus and fallopian tubes should be left in situ because the uterus may be needed later if the patient desires pregnancy through donor oocytes or donor embryos. Using this approach, successful pregnancies have been carried by 46,XY gender-reversed females.16
|
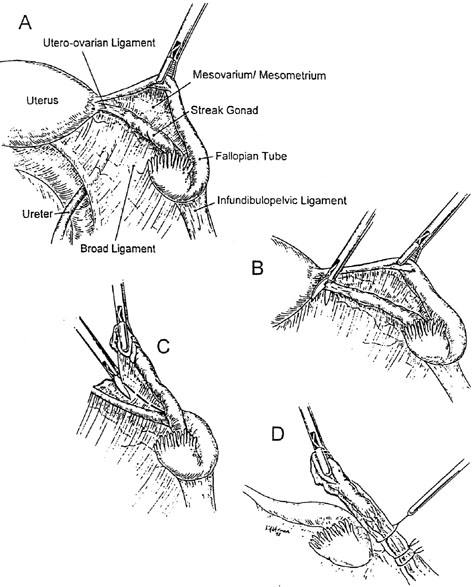
Labeling an individual XY gonadal dysgenesis without somatic anomalies is a diagnosis of exclusion, but most cases will fall into this category. The forms of XY gonadal dysgenesis discussed later in this chapter constitute a minority. DNA perturbations in SRY are not usually detected, and other information (e.g., pedigree) rarely points to a specific etiology. In the majority of cases no etiology is evident using current technologies.
RELATION BETWEEN XY GONADAL DYSGENESIS AND 45,X/46,XY MOSAICISM
XY gonadal dysgenesis depends on exclusion of 45,X cells for its nosologic distinctiveness. This is never certain and often only a single nonreproductive tissue (blood) is studied. No clinical features distinguish XY gonadal dysgenesis from phenotypic females with a 45,X/46,XY complement. Is idiopathic XY gonadal dysgenesis indeed a distinct entity, or could it simply be part of the 45,X/46,XY spectrum? Could ostensible 46,XY cases merely be 45,X/46,XY mosaics in which the monosomic cell line is present only in germ cells or nonhematogenous tissue? Reasons for considering XY gonadal dysgenesis as part of the 45,X/46,XY spectrum include: (1) bilateral streak gonads occur in individuals whose complements may be either 45,X/46,XY or 46,XY; (2) mixed gonadal dysgenesis (streak gonad and contralateral dysgenetic testis) occur in 45,X/46,XY, as well as in individuals whose complements are ostensibly 46,XY; (3) the same rare tumors (gonadoblastomas and dysgerminomas) occur in XY gonadal dysgenesis and 45,X/46,XY; (4) gonadal structures intermediate between those usually present in XY gonadal dysgenesis and those usually present in mixed dysgenesis (45,X/46,XY);17 and (5) 45,X line cells claimed to be present in a gonad of an individual whose sib had XY gonadal dysgenesis.18
Reasons for concluding that idiopathic XY gonadal dysgenesis is different etiologically from 45,X/46,XY mosaicism include the familial aggregates of XY gonadal dysgenesis. 45,X/46,XY mosaicism has not been reported in sibs, although 45,X/46/XY/47,XYY mosaicism was described in sibs whose parents were consanguineous.19, 20 Nonetheless, familial aggregates alone argue strongly against simply considering idiopathic XY gonadal dysgenesis as part of the 45,X/46,XY spectrum. Moreover, undetected monosomic lines would, even if present in XY gonadal dysgenesis, have arisen at a different time and in a different tissue than in individuals in whom 45,X/46,XY mosaicism is demonstrable in blood.
GONADAL DYSGENESIS CAUSED BY PERTURBATIONS OF SRY
Approximately 10–15% of XY gonadal dysgenesis cases show perturbations of SRY.21, 22, 23, 24 SRY consists of two open reading frames, 99 and 273 amino acids in length. The key sequence involves a high mobility group (HMG) box encompassing codons 13–82. This domain box shares characteristics in common with other DNA binding sequences. When perturbation occurs within SRY, it almost always involves the HMG box (Fig. 3). Less commonly, mutations are recognized in other regions of SRY9, 25, 26, 27 or downstream to SRY.28
|
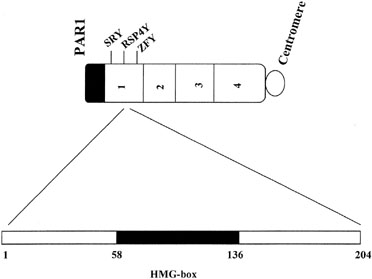
The exact function of SRY remains unclear at the cellular level.24, 29 Presumably SRY binds DNA at a consensus sequence (AATAAC) in all species. The role of SRY in testes differentiation may involve DNA bending,30 perhaps juxtaposing more than one testicular determining genes and, hence, facilitating transcription or interaction between certain gene products. Ostrer discusses this topic in more detail in the chapter "Genetics of Sexual Differentiation", and it has been considered elsewhere by this author.31
X-LINKED RECESSIVE XY GONADAL DYSGENESIS
In multiple kindreds, more than one family member has had XY gonadal dysgenesis. These include concordant monozygotic twins32 and multiple affected sibs of nonconsanguineous parents.18, 33, 34, 35, 36, 37, 38, 39, 40, 41 In four families the trait segregated in the manner expected of an X-linked recessive or male-limited autosomal dominant gene.41, 42, 43, 44, 45 XY gonadal dysgenesis may thus result from an X-linked recessive or male-limited autosomal dominant gene. This mode of action of the gene is uncertain, despite rampant speculation over several decades.
COEXISTENCE OF XY GONADAL DYSGENESIS WITH THE TRIAD OF XY GENITAL AMBIGUITY, BILATERAL TESTES, AND PERSISTENT MULLERIAN DERIVATIVES: PARTIAL GONADAL DYSGENESIS?
Several families have been reported in which at least one sib had XY gonadal dysgenesis, whereas another 46,XY sib had genital ambiguity, bilateral testes, and müllerian derivatives.46, 47, 48 These families were reported decades ago and have not been studied with contemporary methods. However, they are not dissimilar to those described in the well-cited family of Berkovitz and colleagues.49 These authors described eight patients with “46,XY partial gonadal dysgenesis”. The phenotype was typically genital ambiguity or clitoromegaly, a vagina but often partial labioscrotal fusion, müllerian derivatives and gonads that included bilateral or unilateral dysgenetic testes, and often a contralateral streak gonad. Mosaicism (45,X/46,XY) was not detected, but the extent of search may not have been exhaustive.
Ostrer50 reported a four-generation family including seven women with XY gonadal dysgenesis and four men with hypospadia or cryptorchidism. In this family a putative autosomal dominant gene was localized to chromosome 5 by genome-wide linkage analysis. The triad of clinical features listed above is usually not heritable, despite familial aggregates typically existing among male pseudohermophrodites (46,XY), individuals with genital ambiguity, bilateral testes, and no Mullerian derivatives. Potential explanations for these families include: (1) undetected 45,X cells in multiple family members (familial mosaicism) thus explaining the presence of a uterus; (2) the gene that causes XY gonadal dysgenesis (see above) being capable of varied expressivity in families; or (3) a different gene from that controlling XY gonadal dysgenesis exists and is manifesting varied expressivity
DAX-1, Xp21 DUPLICATIONS, AND XY GONADAL DYSGENESIS
Since the report of Bernstein and colleagues51 it has been accepted that XY gender reversal may be associated with duplication of the X short arm (Xp21). Initial observations emphasized not only gender reversal but coexistence of multiple malformations. When these features are combined with XY gender reversal, the name Gardner-Silengo-Wachtel syndrome is applied.51, 52, 53
The phenotypic female proband of Bernstein and colleagues51 showed enlargement of the cranial ventricles, was mentally retarded, and manifested myoclonic jerks. Her height, weight, and head circumference were far below the third centile, and she displayed many dysmorphic features: transparent skin, facial and skull asymmetry, low-set ears, prominent frontal bossing, hypertelorism, downward slanting palpebral fissures, depressed nasal bridge, prognathism, narrow lips, carp-shaped mouth, cleft palate, malaligned teeth, clinodactyly, and prominent hyperextended heels. Internal anomalies included ventricular (cardiac) septal defect, fatty metamorphosis of the liver, and fibrotic gallbladder. Hypercholesterolemia and hyperbilirubinemia were present. External genitalia were female: normal vagina; müllerian derivatives consisted of cervix, uterus, and fallopian tubes; gonads consisted mostly of ovarian stroma but with a few identifiable primordial follicles. Unexpectedly, the proband had one normal Y chromosome and one X chromosome characterized by duplication of a portion of the X short arm (Xp). The identical abnormal X chromosome was present in the proband’s mother, sister, and maternal grandmother. All had a second normal X, and all were phenotypically normal. Amniocentesis in a subsequent pregnancy by the proband’s mother revealed a chromosomal complement identical to that of the proband, for which reason the pregnancy was terminated. The aborted fetus showed many of the features present in the proband: hypertelorism, micrognathia, low-set ears, cleft palate, ventricular septal defect, and prominent heels. External genitalia were again unequivocably female, müllerian derivatives present, and the ovary contained primary follicles. In later cases54 phenotypic abnormalities including gender reversal were confirmed, but adrenal hypoplasia was characteristic. The gene thought to be duplicated was first called dosage-dependent sex reversal (DSSR) and then DAX-1 (dose-sensitive sex reversal-adrenal hypoplasia congenital, X chromosome).
DAX-1 perturbations are not a common explanation for XY gender reversal. Bardoni and colleagues55 studied 27 cases of gender reversal and found only one to have DAX-1 duplication. DAX-1 probably is not the X-linked recessive gene responsible for X-linked XY gonadal dysgenesis (see above). DAX-1 was once postulated to be the primary ovarian determinant, but this now seems less likely.56
Ostrer29 and McElreavey and Fellous24 sought to explain the relation between SRY X loci that play a role in testicular differentiation. One postulate is that SRY directly represses DAX-1, a locus on Xp that otherwise would direct ovarian development. In the presence of duplication of Xp, DAX-1 cannot be suppressed by SRY; thus, ovaries develop. DAX-1 may also be regulated by WNT-4, a locally acting growth factor located on chromosome 1 (1p53). The basis for this hypothesis is that duplication of 1p and overexpression of WNT-4 resulted in upregulation of DAX-1 in a gender-reversed XY female.57 Against the concept that Xp21 duplication suppresses SRY to cause sex reversal is that 47,XXY males fail to show sex reversal. It is possible that X-inactivation exists in XXY but not in Xp duplications, or involves loss or perturbation of a gene contiguous or near DAX-1.
XY GONADAL DYSGENESIS AND AUTOSOMAL DELETIONS: 2q, 9p, 10q
A surprising number of autosomal perturbations are now recognized to be associated with XY gonadal dysgenesis. Even more surprising, affected cases may show no abnormalities other than those involving reproductive organs. In this section only those autosomal deletions having no somatic anomalies will be discussed. Mutations of SOX9, which cause campomelic dwarfism (17q), and the various WT-1 mutations causing Densy-Drash and Frasier syndromes (11p), are discussed separately.
Deletions of 9p, especially 9p24.3, may cause XY gonadal dysgenesis.58, 59, 60, 61, 62, 63 Chromosomal region 9p24.3 contains a domain homologous to key gender-determining genes in Caenorhabditis elegans (mab3) and Drosophila (double sex or dsx). The putative human locus was initially termed DMT, but now is called DMRT1 (double sex and mab3-related transcription factor 1). Ferguson-Smith and colleagues64 concluded that del(9p24.3) was a common cause of 46,XY gonadal dysgenesis in SRY-positive cases after finding that of 11 46,XY females with SRY, three showed complete deletion of one DMT1 allele. More recently this group more precisely identified a submicroscopic deletion that identified the 9p gender-reversal region.65 Raymond and colleagues66 found that 9p24.3 contains two relevant domains: DMRT1 and DMRT2. Sequencing 87 unexplained XY gender-reversal cases revealed only one potential mutation in DMRT1 and none in DMRT2. The same group later found no deletions, as assessed through a DMRT1 fluorescent in situ hybridization (FISH) probe.67 Moreover, the smallest reported region of 9p capable of producing gender reversal showed neither DMRT1 nor DMRT2, suggesting that both genes must be deleted for gender reversal, thus postulating that two genes could imply a quantitative threshold for DMRT gene activity, below which testicular differentiation is impeded. With such a mechanism, phenotype might predictably be expected to vary (varied expressivity), as indeed it does.62 Irrespective, such perturbations seem to be uncommon explanations for XY gonadal dysgenesis, Ottolenghi and colleagues68 found mutation of DMRT2 in sequencing nine 46,XY gender-reversed females.
Deletions of 2q and 10q have also been reported. Slavotinek and colleagues69 reported gender reversal with deletions of 2q33. Waggoner and colleagues70 summarized gender reversal associated with deletion of 10q26; of note the gene for SF-1 (see below) is located on 10q, but not 10q26 (see XY Sex Reversal Caused by Deficiency of Steroidogenic Factor-1).
XY GONADAL DYSGENESIS AND AUTOSOMAL DUPLICATION (1p)
Wieacker and colleagues71 reported XY gender reversal associated with duplication of 1p (p22.3 → p32.2). A later XY gender reversal case caused by 1p duplication was that already alluded to as that of Jordan and colleagues.57 In that case duplication of 1p resulted in overexpression of WNT-4, in turn leading to dysregulation of the X-linked gene DAX1.57
XY GONADAL DYSGENESIS AND WT1 MUTATIONS: DENYS-DRASH AND FRASIER SYNDROMES
Mutations of WT-1 (Wilms’ tumor oncogene) produce a variety of genital abnormalities. Some of which result in 46,XY gender reversal (XY females), whereas others result in male pseudohermaphroditism (genital ambiguity). WT-1 is located on chromosome 11p, and is ten exons in length. At least 32 isoforms exist.24 The association of nephropathy, genital ambiguity, and Wilms’ tumor was first appreciated in a (46,XY) male child, and the disorder became known as Denys-Drash syndrome. This was prior to recognition of the significance of WT-1 perturbations.72 Gessler and colleagues73 then observed mental retardation, aniridia, and Wilms’ tumor in association with deletions of 11p13. This constellation of features proved to be caused by a minute chromosomal deletion (contiguous gene syndrome) that deleted WT-1, resulting in loss of tumor suppressor function.
Gonadal development in Denys-Drash ranges from streak gonads through dysgenetic testes to true hermaphroditism.74 Most cases of Denys-Drash syndrome are males with genital ambiguity, but a few 46,XY cases are phenotypically female. Approximately, half the affected phenotypic females are 46,XY.75 The genetic mechanism involves production of a mutant gene product that exerts a dominant negative effect, leading to loss of tumor suppressor and interference with testicular differentiation.
Frasier syndrome is caused by a different perturbation of WT-1. In this condition renal parenchymal disease and XY gender reversal occurs.76 The case of XY gonadal dysgenesis gonadoblastoma and renal parenchymal failure described by Simpson and colleagues77 represented what is now called Frasier syndrome. Frasier syndrome has been reported in a 46,XX women whose 46,XY sib was also affected.78 Both had characteristic focal segmental glomerulosclerosis, but only the XY sib had gonadal abnormalities. That sib had prototypic XY gonadal dysgenesis and female external genitalia; the XX sib had normal ovaries. The WT-1 mutation that causes Frasier syndrome thus affects testicular but not ovarian differentiation.
Frasier syndrome and Denys-Drash syndrome are caused by different types of perturbation involving WT-1.79 The latter is caused by deletion of WT-1. The former is caused by an altered ratio of a specific splicing of amino acids. WT-1 has several alternate translation initiation sites, alternate differential. An alternate donor acceptor site at the exon 9 boundary results in differential splicing of the three amino acids lysine (K), threonine (T), and serine (S).79, 80 The ratio of KTS(+) to non-KTS(−) transcripts is apparently pivotal, differing between testes and ovary.81 Perturbation of the ratio is pivotal in causing Frasier syndrome.
In mice, the WT-1 null mutation results in failure of either sex to develop gonads (and kidneys).82 Both SRY and WT-1 are found in the testes, with the latter evident earlier. For this reason it has been proposed that WT-1 is required upstream of SRY (i.e., before SRY is expressed).24
XY GONADAL DYSGENESIS AND CAMPOMELIC DYSPLASIA (SOX9)
Campomelic dysplasia is a well-characterized multiple malformation syndrome distinguished by bowing of long bones, abnormal facies, and other skeletal anomalies (Fig. 4). The gene causing this disorder is located on 17q 24.3–q25.1. Its perturbation involves the DNA binding protein SOX9.83, 84
Fig. 4. Campomelic dysplasia, often caused by perturbation of the SOX9 gene on 17q. (Jones KL: Smith’s Recognizable Patterns of Human Malformation, p. 345. 5th ed. 1997.) |
Deletion of 17q involving SOX9 may cause both campomelic dysplasia and XY gonadal dysgenesis (gender reversal).85, 86 The precise mode of action is still somewhat unclear but more than simply the SOX9 locus may be involved.
Olney and colleagues87 reported interstitial deletion of 17q 23.3q-24.3 in complete absence of SOX9 and campomelic dysplasia, suggesting haplo-insufficiency as the molecular mechanism. SOX9 mutations do not, however, always manifest as XY gender reversal, nor do mutations in SOX9 produce 46,XY gonadal dysgenesis in the absence of skeletal anomalies.88 This may reflect modifying genes or mutations in the long (1 mb) SOX9 promotor region.89 Other observations indicate 17q is a pivotal autosomal region for control of sexual differentiation in general. Huang and colleagues90 reported duplication of 17p associated with presumptive bilateral testes and incomplete male-like external genitalia. In mice insertional mutagenesis of a gene (odd sex, or ods) upstream of SOX9 results in unscheduled testicular development in XX animals.91 It has thus been speculated that SRY (murine SRY) ordinarily derepresses this region in order to permit testicular development in XY embryos; a mutation resulting in unexpected derepression would result in the same phenotype in XX embryos. Indeed, Vidal and colleagues92 created XX transgenic mice that expressed SOX9 and showed testes despite lack of Sry.
XY GONADAL DYSGENESIS AND ALPHA-THALASSEMIA (ATX)
The X–linked gene ATXC causes mental retardation, α-thalassemia, abnormal facial features (upturned nose, carp-shaped mouth), and male pseudohermaphrodies. The reproductive phenotype extends to gender-reversed (XY) females.93, 94, 95, 96 The gene designation ATX is derived from (α-thalassemia X chromosome). This gene is located on Xq12–q21.31. A member of the DNA helicase family, more than 25 mutations are reported.97, 98
XY GONADAL DYSGENESIS IN OTHER MALFORMATION SYNDROMES
Several multiple malformation syndromes characterized or associated with XY gender reversal have been discussed here. We have already mentioned Gardner-Silengo-Wachtel syndrome,54 Frasier and Denys-Drash syndromes, and compomelic dysplasia. Far more often genital ambiguity occurs in 46,XY cases with multiple malformations, nosologically placing these cases as male pseudohermaphrodites.
Other multiple malformation syndromes in which external genitalia are female indicate: (1) XY gonadal dysgenesis and ectodermal anomalies99 and (2) XY gonadal dysgenesis, spastic paraplegia, optic atrophy, and microcephaly.100 Both have been reported in only single families. XY gonadal dysgenesis (gender-reversed fetus) has also been observed in the presence of holoprosencephaly.101
XY GENDER REVERSAL CAUSED BY LHR MUTATIONS (LEYDIG CELL AGENESIS)
If embryonic testes lack Leydig cells, testosterone and, hence, dihydrotestosterone are not synthesized; thus, embryos will show female external genitalia and no uterus. Embryonic testes presumably secrete AMH, predictably explaining absence of a uterus as predicted in 46,XY individuals. As adults, LH is elevated.
Autosomal recessive inheritance has long been accepted on the basis of affected sibs and parental consanguinity. The molecular basis involves mutation in the LHR gene, located on chromosome 2.102 Thus, Leydig cells fail to develop because LH has inadequate receptor to exert its normal effect during embryogenesis, resulting in inadequate virilization and failure of gonadal differentiation. Complete resistance to LH produces XY females, whereas partial resistance leads to males with a small penis or hypospadias. Various deletions, missense mutations, and nonsense mutations (stop codons) have been recognized.103, 104, 105
The LHR gene consists of 11 exons and 699 amino acids. Intracellular domains, intracellular and extracellular loops, transmembrane domains, and extracellular domains exist. A dozen different LHR mutations (molecular heterogeneity) have been found in 46,XY females with Leydig cell hypoplasia. In some reports 46,XX sibs102 were similar in phenotype to their 46,XY sibs. Kremer and colleagues106 reported two 46,XX sibs of consanguineous parents; homozygosity existed for a missense mutation (C593R).
XY GENDER REVERSAL CAUSED BY DEFICIENCY OF STEROIDOGENIC FACTOR-1
Steroidogenic factor-1 appears to play a pivotal, albeit still poorly defined, role in the hypothalamic–pituary gonadal axis. The SF-1 gene product is encoded by the gene FTZ1, located on human 9q33. FTZ1 is a zinc finger protein. Because SF-1 has no known ligand, it is designated as an orphan nuclear receptor. In mice, FTZ1 gene knockouts result in both adrenal and gonadal disturbances, as well as abnormalities of the hypothalamus and pituitary gonadatropes.107, 108 Although the role of SF-1 in these interactions is not clear, mutations in human FTZ1-disturbing SF-1 receptors would be expected to cause significant abnormalities of sexual development.
One human case involving a SF-1 mutation has been reported.56 This 46,XY individual showed primary adrenal failure, female external genitalia, streak gonads, and normal müllerian derivatives. SRY, StAR, and DAX1 were normal. A 2-base pair substitution was found in codon 35 (G35E), resulting in a mutated glycine. This is the last amino acid of the first zinc finger of SF-1, a finding suggesting perturbation of DNA binding. The mutation on the other allele, presumably necessary to explain the phenotype, was not detected.
The presence of a uterus supports the hypothesis that SF-1 regulates repression of AMH. Thus, a role for SF-1 in this regard was incorporated into Figure 1.
DEFICIENCY OF 17α-HYDROXYLASE/ 17,20-DESMOLASE (CYP17)
The bifunctional enzyme 17α-hydroxylase/17,20 desmolase (CYP17) governs two conversions in the adrenal and gonadal biosynthetic pathway (Fig. 5). If the enzyme is deficient, neither androgens nor estrogens are properly synthesized. Affected 46,XX individuals may thus present with primary amenorrhea and hypergonadotropic hypogonadism. Most of the 150 reported cases of 17α-hydroxylase deficiency are male (46,XY) and show (male) genital ambiguity. However, 46,XY cases presenting with female phenotype exist.
|
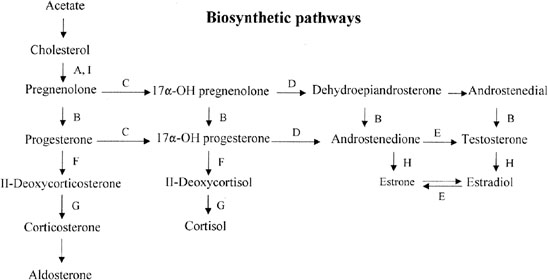
An important diagnostic clue is hypertension, the pathogenesis of which is hypervolemia secondary to mineralocorticoid excess. The clinical presentation is otherwise indistinguishable from XX gonadal dysgenesis without somatic anomalies. Diagnosis of CYP17 deficiency is based on elevated progesterone, deoxycorticosterone, and corticosterone coupled with decreased testosterone and estrogen. Treatment involves control administrative.
Females (46,XX) deficient for 17α-hydroxylase/17,20-desmolase (CYP17) show hypoplastic ovaries that are sometimes streak-like in appearance; oocytes appear incapable of reaching diameters greater than 2.5 mm.109 However, exogenous gonadotropin stimulation can produce oocytes suitable for in vitro fertilization (IVF).110
17α-Hydroxylase deficiency is inherited in autosomal recessive fashion. The gene (CYP17) is located on 10q24-25, and the gene product is a cytochrome P450 enzyme. This single gene (and enzyme) is responsible for both 17α-hydroxylase and 17,20-desmolase (lyase) actions.
Molecular
More than 20 different mutations have been identified in CYP17, scattered among the eight exons (Fig. 6). Mutations include missense mutations, duplications, deletions, and premature protein truncation.111 Most mutations have been observed in only a single family, yet another example of molecular hetereogeneity. An exception exists in Mennonites of Dutch origin, where a 4-base duplication in exon 8 accounts for most cases.112 This founder mutation originated in Friesland.
Few patients having deficiency of both 17α-hydroxylase and 17,20 lyase activities have been analyzed, but mutations different from those only showing deficient 17α-hydroxylase activity have been found.113, 114 Transfection experiments show that only 5% of 17α-hydroxylase activity is sufficient for the estrogen production necessary for normal secondary sexual characteristics in a 46,XX individual; however, 25% of enzyme activity is necessary to virilize external genitalia in males.111, 113 Targeted mutagenesis in the rat gene indicates that mutations closer to the 5` end are more deleterious (see Fig. 6).
|
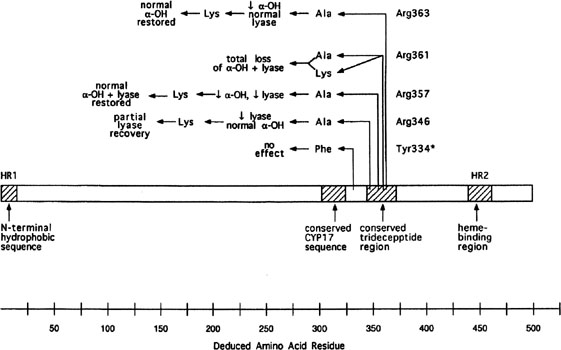
OTHER GENES IMPLICATED IN TESTICULAR DIFFERENTIATION
Several other genes have received attention on the basis of animal models showing phenotypes of relevance. Few studies involving humans have been reported.
LHX8 (Lim) is a homeobox gene located on the 1q25-31, expressed around the time of formation of gonads. No mutations were found in 41 cases of XY gonadal dysgenesis by Ottolenghi et al.;115 the sample included two XY sibs.
EMX2 is a homeobox gene expressed in the developing head and urogenital system. No genital abnormalities were observed in two brothers with schizencephaly (cleft within cerebral hemisphere), each of whom had a heterozygous mutation.116
m33 gene mutation in mice modifies chromatin structure. Perturbations can cause murine XY sex reversal.117 No human studies have been reported.
Fgf9 is involved in Sertoli cell development and mesonephic migration.118
Desert Hedge Hog (DHH) is a signaling protein localized to 12q. One 46,XY phenotypic female with minifasicular neuropathy showed female external genitalia; a testis was present on one side and a streak gonad on the other.119 A homozygous mutation involving the initiating condon of exon 1 resulted in no gene product.
REFERENCES
Jost A: Problems of fetal endocrinology. The gonadal and hypophyseal hormones. Rec Prog Horm Res 8:379, 1953 |
|
Simpson JL: Disorders of the gonads, genital tract, and genitalia. In Rimoin DL, Connor JM, Pyeritz RE (eds): Principles and Practice of Medical Genetics. pp. 2055-2092, 5th ed. New York, Churchill-Livingstone, 2007 |
|
Simpson JL: Disorders of abnormal sexual differentiation. (In) pediatric and Adolescent Gynecology, 3rd Edition (JS Sanfilippo, D Muram, PA Lee, J Dewhurst ed). Informa Healthcare, New York, NY, In press. |
|
Patsavoudi E, Magre S, Castanier M et al. Dissociation between testicular morphogenesis and functional differentiation of Leydig cells. J Endocrinol 105:235, 1985 |
|
Magre S, Jost A: Dissociation between testicular organogenesis and endocrine cytodifferentiation of Sertoli cells. Proc Natl Acad Sci USA 81:7831, 1984 |
|
Behringer RR, Cate RL, Froelick GJ et al. Abnormal sexual development in transgenic mice chronically expressing mullerian inhibiting substance. Nature 345:167, 1990 |
|
Yu RN, Ito M, Saunders TL et al. Role of AHCH in gonadal development and gametogenesis. Nat Genet 20:353, 1998 |
|
Skre H, Bassoe HH, Berg K et al. Cerebellar ataxia and hypergonadotropic hypogonadism in two kindreds. Chance concurrence, pleiotropism or linkage? Clin Genet 9:234, 1976 |
|
Brown S, Yu C, Lanzano P et al: A de novo mutation (Gln2Stop) at the 5&vprime; end of the SRY gene leads to sex reversal with partial ovarian function. Am J Hum Genet 62:189, 1998 |
|
Jirasek J. Principles of reproductive embryology. In Simpson JL (ed): Disorders of Sexual Differentiation: Etiology and Clinical Delineation. pp. 51, 110 San Diego, Academic Press, 1976 |
|
Cussen LJ, MacMahon RA. Germ cells and ova in dysgenetic gonads of a 46-XY female dizygotic twin. Am J Dis Child 133:373, 1979 |
|
Swyer GIM. Male pseudohermaphroditism: A hitherto underscribed form. BMJ 2:709, 1955 |
|
Simpson JL, Photopulos G: The relationship of neoplasia to disorders of abnormal sexual differentiation. Birth Defects Orig Artic Ser 12(1):15, 1976 |
|
Wilson EE, Vuitch F, Carr BR: Laparoscopic removal of dysgenetic gonads containing a gonadoblastoma in a patient with Swyer syndrome. Obstet Gynecol 79:842, 1992 |
|
Pisarska MD, Simpson JL, Zepeda DE et al: Laparoscopic removal of streak gonads in 46,XY or 45,X/46,XY gonadal dysgenesis. J Gynecol Tech 4:95, 1998 |
|
Kan AK, Abdalla HI, Oskarsson T: Two successful pregnancies in a 46,XY patient. Hum Reprod 12:1434, 1997 |
|
Fraccaro M, Lindsten J, Klinger HP et al: Cytogenetical and clinical investigations in four subjects with anomalies of sexual development. Ann Hum Genet 29:281, 1966 |
|
Cohen MM, Shaw MW: Two XY siblings with gonadal dysgenesis and a female phenotype. N Engl J Med 272:1083, 1965 |
|
Goldstein DP, Lamb EJ: Arrhenoblastoma in first cousins. Report of 2 cases Obstet Gynecol 35:444, 1970 |
|
Hsu LY, Hirschhorn K, Goldstein A et al: Familial chromosomal mosaicism, genetic aspects. Ann Hum Genet 33:343, 1970 |
|
Berta P, Hawkins JR, Sinclair AH et al: Genetic evidence equating SRY and the testis-determining factor. Nature 348:448, 1990 |
|
Pivnick EK, Wachtel S, Woods D et al: Mutations in the conserved domain of SRY are uncommon in XY gonadal dysgenesis. Hum Genet 90:308, 1992 |
|
Cameron FJ, Sinclair AH: Mutations in SRY and SOX9: Testis-determining genes. Hum Mutat 9:388, 1997 |
|
McElreavey K, Fellous M: Sex determination and the Y chromosome. Am J Med Genet 89:176, 1999 |
|
Tajima T, Nakae J, Shinohara N et al: A novel mutation localized in the 3' non-HMG box region of the SRY gene in 46,XY gonadal dysgenesis. Hum Mol Genet 3:1187, 1994 |
|
Veitia R, Ion A, Barbaux S, et al. Mutations and sequence variants in the testis-determining region of the Y chromosome in individuals with a 46,XY female phenotype. Hum Genet 99:648, 1997 |
|
McElreavey K, Rappaport R, Vilain E, et al. A minority of 46,XX true hermaphrodites are positive for the Y-DNA sequence including SRY. Hum Genet 90:121, 1992 |
|
McElreavey K, Vilain E, Barbaux S et al: Loss of sequences 3&vprime; to the testis-determining gene, SRY, including the Y pseudoautosomal boundary associated with partial testicular determination. Proc Natl Acad Sci USA 93:8590, 1996 |
|
Ostrer H: Sexual differentiation. Semin Reprod Med 18:41, 2000 |
|
Giese K, Pagel J, Grosschedl R: Distinct DNA-binding properties of the high mobility group domain of murine and human SRY sex-determining factors. Proc Natl Acad Sci USA 91:3368, 1994 |
|
Simpson JL, Elias S: Genetics in Obstetrics and Gynecology. 3rd ed., Philadelphia, W.B. Saunders, 2003 |
|
Frasier SD, Bashore RA, Mosier HD: Gonadoblastoma associated with pure gonadal dysgenesis in monozygous twins. J Pediatr 64:740, 1964 |
|
Brogger A, Strand A: Contribution to the study of the so-called pure gonadal dysgenesis. Acta Endocrinol 48:490, 1965 |
|
Stanesco V, Maximilian C, Florea I et al: [3 sisters with pure gonadal dysgenesis and XY karyotype]. Ann Endocrinol 29:449, 1968 |
|
Boczkowski K: Sex determination and gonadal differentiation in man. A unifying concept of normal and abnormal sex development Clin Genet 2:379, 1971 |
|
Talerman A: Gonadoblastoma and dysgerminoma in two siblings with dysgenetic gonads. Obstet Gynecol 38:416, 1971 |
|
Berger R, Binoux M, Chassan E, and colleagues: [Pure familial gonadal dysgenesis]. Ann Endocrinol 33:35, 1972 |
|
Campodonico I, Venegas E, Saito E et al: [Swyer’s syndrome (pure gonadal dysgenesis) and gonadal neoplasias in 3 sisters]. Rev Child Obstet Ginecol 38:155, 1973 |
|
Anderson CT Jr, Carlson IH: Elevated plasma testosterone and gonadal tumors in two 46XY “sisters”. Arch Pathol 99:360, 1975 |
|
Fleishman PR, Berlin M, Dana MM: Gonadal dysgenesis in two siblings. Am J Obstet Gynecol 124:208, 1976 |
|
Mann JR, Corkery JJ, Fisher HJ et al: The X linked recessive form of XY gonadal dysgenesis with a high incidence of gonadal germ cell tumours: clinical and genetic studies. J Med Genet 20:264, 1983 |
|
Sternberg WH, Barclay DL, Kloepfer HW: Familial XY gonadal dysgenesis. N Engl J Med 278:695, 1968 |
|
Espiner EA, Veale AM, Sands VE et al: Familial syndrome of streak gonads and normal male karyotype in five phenotypic females. N Engl J Med 283:6, 1970 |
|
Simpson JL: Disorders of Sexual Differentiation: Etiology and Clinical Delineation. New York, Academic Press, 1976 |
|
German J, Simpson JL, Chaganti RS et al: Genetically determined sex-reversal in 46,XY humans. Science 202:53, 1978 |
|
Baron J, Rucki T, Simm S: Familial gonadal malformations. Gynaecologia 153:298, 1962 |
|
Barr ML, Carr DH, Plunkett ER et al: Male pseudohermaphroditism and pure gonadal dysgenesis in sisters. Am J Obstet Gynecol 99:1047, 1967 |
|
Chemke J, Carmichael R, Stewart JM et al: Familial XY gonadal dysgenesis. J Med Genet 7:105, 1970 |
|
Berkovitz GD, Fechner PY, Zacur HW et al: Clinical and pathologic spectrum of 46,XY gonadal dysgenesis: Its relevance to the understanding of sex differentiation. Medicine 70:375, 1991 |
|
Ostrer H, Jawaheer D, Juo S-H et al: A gene for human testis determination maps to chromosome 5. Am J Hum Genet (Supplement) 69:213, 2001 |
|
Bernstein R, Koo GC, Wachtel SS: Abnormality of the X chromosome in human 46,XY female siblings with dysgenetic ovaries. Science 207:768, 1980 |
|
Wolman SR, McMorrow LE, Roy S et al: Aberrant testicular differentiation in 46,XY gonadal dysgenesis: Morphology, endocrinology, serology. Hum Genet 55:321, 1980 |
|
Simpson JL: Genetic heterogeneity in XY sex reversal. Potential pitfalls in isolating the Testis-Determining-Factor (TDF) In Wachtel S (ed): Evolutionary Mechanisms in Sex Determination. pp. 265, 277 Baton Rouge, CRC Press, 1989 |
|
Greenberg F, Gresik MV, Carpenter RJ et al: The Gardner-Silengo-Wachtel or genito-palato-cardiac syndrome: Male seudohermaphroditism with micrognathia, cleft palate, and conotruncal cardiac defect. Am J Med Genet 26:59, 1987 |
|
Bardoni B, Zanaria E, Guioli S et al: A dosage sensitive locus at chromosome Xp21 is involved in male to female sex reversal. Nat Genet 7:497, 1994 |
|
Achermann JC, Ito M, Ito M, et al: A mutation in the gene encoding steroidogenic factor–1 causes XY sex reversal and adrenal failure in humans. Nat Genet 22:125, 1999 |
|
Jordan BK, Mohammed M, Ching ST et al: Up-regulation of WNT-4 signaling and dosage-sensitive sex reversal in humans. Am J Hum Genet 68:1102, 2001 |
|
Bennett CP, Docherty Z, Robb SA et al: Deletion 9p and sex reversal. J Med Genet 30:518, 1993 |
|
McDonald MT, Flejter W, Sheldon S et al: XY sex reversal and gonadal dysgenesis due to 9p24 monosomy. Am J Med Genet 73:321, 1997 |
|
Veitia R, Nunes M, Brauner R et al: Deletions of distal 9p associated with 46,XY male to female sex reversal: definition of the breakpoints at 9p23.3-p24.1 Genomics 41:271, 1997 |
|
Flejter WL, Fergestad J, Gorski J, et al: A gene involved in XY sex reversal is located on chromosome 9, distal to marker D9S1779. Am J Hum Genet 63:794, 1998 |
|
Veitia RA, Nunes M, Quintana-Murci L, et al: Swyer syndrome and 46,XY partial gonadal dysgenesis associated with 9p deletions in the absence of monosomy-9p syndrome. Am J Hum Genet 63:901, 1998 |
|
Guioli S, Schmitt K, Critcher R, et al: Molecular analysis of 9p deletions associated with XY sex reversal: Refining the localization of a sex-determining gene to the tip of the chromosome. Am J Hum Genet 63:905, 1998 |
|
Ferguson-Smith MA, Sanoudou D, Lee C: Microdeletion of DMT1 at 9p24,3 is the commonest cause of 46,XY females. Am J Hum Genet 63:A162, 1998 |
|
Calvari V, Bertini V, De Grandi A, et al: A new submicroscopic deletion that refines the 9p region for sex reversal. Genomics 65:203, 2000 |
|
Raymond CS, Parker ED, Kettlewell JR, et al: A region of human chromosome 9p required for testis development contains two genes related to known sexual regulators. Hum Mol Genet 8:989, 1999 |
|
Mengelt AM, Raymond CS, Brown LG, et al: FISH screening for microdeletions of DMRT1 in 46,XY sex reversed individuals. Am J Hum Genet 65:A351, 1999 |
|
Ottolenghi C, Veitia R, Barbieri M, et al: The human doublesex-related gene, DMRT2, is homologous to a gene involved in somitogenesis and encodes a potential bicistronic transcript. Genomics 64:179, 2000 |
|
Slavotinek A, Schwarz C, Getty JF, et al: Two cases with interstitial deletions of chromosome 2 and sex reversal in one. Am J Med Genet 86:75, 1999 |
|
Waggoner DJ, Chow CK, Dowton SB, et al: Partial monosomy of distal 10q: three new cases and a review. Am J Med Genet 86:1, 1999 |
|
Wieacker P, Missbach D, Jakubiczka S, et al: Sex reversal in a child with the karyotype 46,XY, dup (1) (p22.3p32.3) Clin Genet 49:271, 1996 |
|
Drash A, Sherman F, Hartmann WH, et al: A syndrome of pseudohermaphroditism, Wilms’ tumor, hypertension, and degenerative renal disease. J Pediatr 76:585, 1970 |
|
Gessler M, Thomas GH, Couillin P, et al: A deletion map of the WAGR region on chromosome 11. Am J Hum Genet 44:486, 1989 |
|
Edidin DV: Pseudohermaphroditism, glomerulopathy, and Wilms tumor (Drash syndrome). J Pediatr 107:988, 1985 |
|
Mueller RF: The Denys-Drash syndrome. J Med Genet 31:471, 1994 |
|
Fraiser JE, Andres GA, Cooney DR: A syndrome of pure gonadal dysgenesis: gonadoblastoma, Wilm’s tumor and nephron disease. Lab Invest 48:4, 1964 |
|
Simpson JL, Chaganti RS, Mouradian J, et al: Chronic renal disease, myotonic dystrophy, and gonadoblastoma in XY gonadal dysgenesis. J Med Genet 19:73, 1982 |
|
Demmer L, Primack W, Loik V, aet al: Frasier syndrome: A cause of focal segmental glomerulosclerosis in a 46,XX female. J Am Soc Nephrol 10:2215, 1999 |
|
Barbaux S, Niaudet P, Gubler MC, et al: Donor splice-site mutations in WT1 are responsible for Frasier syndrome. Nat Genet 17:467, 1997 |
|
Scharnhorst V, Dekker P, van der Eb AJ, et al: Internal translation initiation generates novel WT1 protein isoforms with distinct biological properties. J Biol Chem 274:23456, 1999 |
|
Nachtigal MW, Hirokawa Y, Enyeart-VanHouten DL, et al: Wilms’ tumor 1 and Dax-1 modulate the orphan nuclear receptor SF-1 in sex-specific gene expression. Cell 93:445, 1998 |
|
Kreidberg JA, Sariola H, Loring JM, et al: WT-1 is required for early kidney development. Cell 74:679, 1993 |
|
Sudbeck P, Schmitz ML, Baeuerle PA, et al: Sex reversal by loss of the C-terminal transactivation domain of human SOX9. Nat Genet 13:230, 1996 |
|
Kwok C, Weller PA, Guioli S, et al: Mutations in SOX9, the gene responsible for Campomelic dysplasia and autosomal sex reversal. Am J Hum Genet 57:1028, 1995 |
|
Foster JW, Dominguez-Steglich MA, Guioli S, et al: Campomelic dysplasia and autosomal sex reversal caused by mutations in an SRY-related gene. Nature 372:525, 1994 |
|
Wagner T, Wirth J, Meyer J, et al: Autosomal sex reversal and campomelic dysplasia are caused by mutations in and around the SRY-related gene SOX9. Cell 79:1111, 1994 |
|
Olney PN, Kean LS, Graham D, et al: Campomelic syndrome and deletion of SOX9. Am J Med Genet 84:20, 1999 |
|
Kwok C, Tyler-Smith C, Mendonca BB, et al: Mutation analysis of the 2 kb 5' to SRY in XY females and XY intersex subjects. J Med Genet 33:465, 1996 |
|
Pfeifer D, Kist R, Dewar K, et al: Campomelic dysplasia translocation breakpoints are scattered over 1 Mb proximal to SOX9: Evidence for an extended control region. Am J Hum Genet 65:111, 1999 |
|
Huang B, Wang S, Ning Y, et al: Autosomal XX sex reversal caused by duplication of SOX9. Am J Med Genet 87:349, 1999 |
|
Bishop CE, Whitworth DJ, Qin Y, et al: A transgenic insertion upstream of sox9 is associated with dominant XX sex reversal in the mouse. Nat Genet 26:490, 2000 |
|
Vidal VP, Chaboissier MC, de Rooij DG, and colleagues: Sox9 induces testis development in XX transgenic mice. Nat Genet 28:216, 2001 |
|
Gibbons RJ, Suthers GK, Wilkie AO, and colleagues: X-linked alpha-thalassemia/mental retardation (ATR-X) syndrome: Localization to Xq12-q21.31 by X inactivation and linkage analysis Am J Hum Genet 51:1136, 1992 |
|
Reardon W, Gibbons RJ, Winter RM, and colleagues: Male pseudohermaphroditism in sibs with the alpha-thalassemia/mental retardation (ATR-X) syndrome. Am J Med Genet 55:285, 1995 |
|
McPherson EW, Clemens MM, Gibbons RJ, and colleagues: X-linked alpha-thalassemia/mental retardation (ATR-X) syndrome: A new kindred with severe genital anomalies and mild hematologic expression. Am J Med Genet 55:302, 1995 |
|
Gibbons RJ, Picketts DJ, Villard L, and colleagues: Mutations in a putative global transcriptional regulator cause X-linked mental retardation with alpha-thalassemia (ATR-X syndrome). Cell 80:837, 1995 |
|
Gibbons RJ, Brueton L, Buckle VJ, and colleagues: Clinical and hematologic aspects of the X-linked alpha-thalassemia/mental retardation syndrome (ATR-X). Am J Med Genet 55:288, 1995 |
|
Gibbons RJ, Bachoo S, Picketts DJ, and colleagues: Mutations in transcriptional regulator ATRX establish the functional significance of a PHD-like domain. Nat Genet 17:146, 1997 |
|
Brosnan PG, Lewandowski RC, Toguri AG, and colleagues: A new familial syndrome of 46,XY gonadal dysgenesis with anomalies of ectodermal and mesodermal structures. J Pediatr 97:586, 1980 |
|
Teebi AS, Miller S, Ostrer H, and colleagues: Spastic paraplegia, optic atrophy, microcephaly with normal intelligence, and XY sex reversal: A new autosomal recessive syndrome? J Med Genet 35:759, 1998 |
|
Witters I, Moerman P, Muenke M, and colleagues: Semilobar holoprosencephaly in a 46,XY female fetus. Prenat Diagn 21:839, 2001 |
|
Sultan C, Lumbroso S: LH receptor defects. In Kempers RD, Cohen J, Haney AF, Younger JB (eds): Fertillity and Reproductive Medicine. Proceedings of the XVI World Congress on Fertility and Sterility. pp. 769, Amsterdam, Elsevier Science, 1998 |
|
Latronico AC, Chai Y, Arnhold IJ, and colleagues: A homozygous microdeletion in helix 7 of the luteinizing hormone receptor associated with familial testicular and ovarian resistance is due to both decreased cell surface expression and impaired effector activation by the cell surface receptor. Mol Endocrinol 12:442, 1998 |
|
Laue LL, Wu SM, Kudo M, and colleagues: Compound heterozygous mutations of the luteinizing hormone receptor gene in Leydig cell hypoplasia. Mol Endocrinol 10:987, 1996 |
|
Latronico AC, Anasti J, Arnhold IJ, and colleagues: Brief report: Testicular and ovarian resistance to luteinizing hormone caused by inactivating mutations of the luteinizing hormone-receptor gene. N Engl J Med 334:507, 1996 |
|
Kremer H, Kraaij R, Toledo SP, and colleagues: Male pseudohermaphroditism due to a homozygous missense mutation of the luteinizing hormone receptor gene. Nat Genet 9:160, 1995 |
|
Parker KL, Schimmer BP: Steroidogenic factor 1: A key determinant of endocrine development and function. Endocr Rev 18:361, 1997 |
|
Luo X, Ikeda Y, Parker KL: A cell-specific nuclear receptor is essential for adrenal and gonadal development and sexual differentiation. Cell 77:481, 1994 |
|
Araki S, Chikazawa K, Sekiguchi I, and colleagues: Arrest of follicular development in a patient with 17 alpha-hydroxylase deficiency: Folliculogenesis in association with a lack of estrogen synthesis in the ovaries. Fertil Steril 47:169, 1987 |
|
Rabinovici J, Blankstein J, Goldman B, and colleagues: In vitro fertilization and primary embryonic cleavage are possible in 17 alpha-hydroxylase deficiency despite extremely low intrafollicular 17 beta-estradiol. J Clin Endocrinol Metab 68:693, 1989 |
|
Yanase T: 17alpha-Hydroxylase/17,20-lyase defects. J Steroid Biochem Mol Biol 53:153, 1995 |
|
Imai T, Yanase T, Waterman MR, and colleagues: Canadian Mennonites and individuals residing in the Friesland region of The Netherlands share the same molecular basis of 17 alpha-hydroxylase deficiency. Hum Genet 89:95, 1992 |
|
Miura K, Yasuda K, Yanase T, and colleagues: Mutation of cytochrome P-45017 alpha gene (CYP17) in a Japanese patient previously reported as having glucocorticoid-responsive hyperaldosteronism: With a review of Japanese patients with mutations of CYP17. J Clin Endocrinol Metab 81:3797, 1996 |
|
Kaneko S, Oshio S, Kobayashi T, and colleagues: Human X- and Y-bearing sperm differ in cell surface sialic acid content. Biochem Biophys Res Commun 124:950, 1984 |
|
Ottolenghi C, Moreira-Filho C, Mendonca BB, et al. Absence of mutations involving the LIM homeobox domain gene LHX9 in 46,XY gonadal agenesis and dysgenesis. J Clin Endocrinol Metab. 86(6):2465-9, 2001. |
|
Failla A, Brunelli S, Granata T et al. A number of schizencephaly patients including 2 brother are heterozygous for germline mutations int he homeobox gene EMX2. Eur J Hum Genet. 5(4); 186-90, Jul-Aug 1997. |
|
Katoh-Fukui Y, Tsuchiya R, Shiroishi T, et al. Male-to-female sex reversal in M33 mutant mice. Nature, 393(6686):688-92, 1998. |
|
Colvin JS, Green RP, Schmahl J, Capel B, Ornitz DM. Male-to-female sex reversal in mice lacking fibroblast growth factor 9. Cell. 104(6):875-89, Mar 2001. |
|
Umehara F, Tate G, Itoh K et al. A novel mutation of desert hedgehog in a patient with 46,XY partial gonadal dysgenesis accompanied by minifascicular neuropathy. Am J Hum Genet.67(5): 1302-5,Nov 2000. |